The deMon2k Home PageHome | Program | Download | Support | Online Manual | Tutorials | News & Events | Publications |Mailing List| Developers |
|||
| Program :: Program description | Features of deMon2k | deMon2k citations | Program branches | History |
The deMon2k Home PageHome | Program | Download | Support | Online Manual | Tutorials | News & Events | Publications |Mailing List| Developers |
|||
| Program :: Program description | Features of deMon2k | deMon2k citations | Program branches | History |
Program description |
|
deMon (density of Montréal) is a software package for density functional theory (DFT) [1-3] calculations. It uses the linear combination of Gaussian-type orbital (LCGTO) approach for the self-consistent solution of the Kohn-Sham (KS) DFT equations. The calculation of the four-center electron repulsion integrals is avoided by introducing an auxiliary function basis for the variational fitting of the Coulomb potential [4-6].
Program features |
|
Additionally, several other features are implemented in private versions of deMon2k. Some of these features are:
Required deMon2k citations |
|
Any publication reporting results obtained with the deMon2k program must include, on depending of the used version of the software, one of the citations listed below.
Any reference to the deMon2k code should include the appropriate release version information.
In addition, all specific program functionalities, exchange-correlation functionals, basis sets and auxiliary function sets must also be cited appropriately, in case they have been used.
Program branches and related programs |
|
The deMon2k program is developed by scientific groups all over the world. The current master version of deMon2k is described and can be downloaded from this web site. However, there exist several deMon2k branches and programs related to deMon2k, which have a different or extended functionality and might be based on an older master version. Please, see the following links for details and more information about functionality and reference to a deMon2k version number:
History |
|
The first widely available version of deMon [7] appeared in 1992. deMon stands for densité de Montréal. The unusual capitalization emphasizes the roots of the code at the Université de Montréal (UdM) simultaneously by its implicit separation into two words [de-Mon(réal)] and by its resemblance to the main edifice of UdM with its tall central tower.
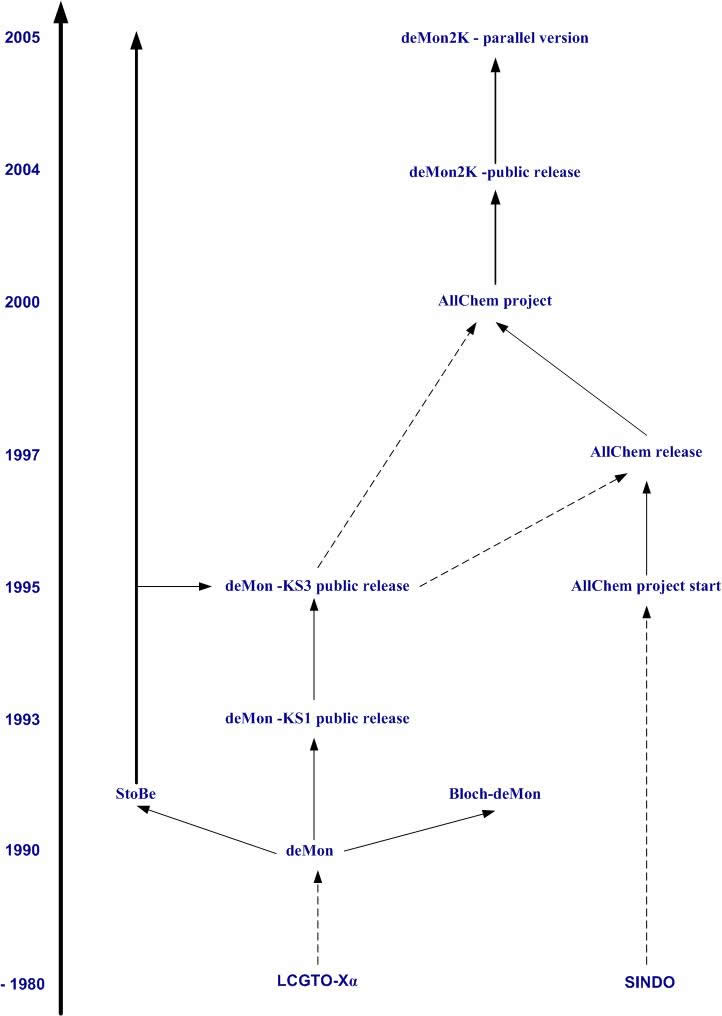
One way to understand deMon is to look at it in its historical context [8]. During the 1970's Slater's X![]() method had been tried and abandoned by quantum chemists. Part of the difficulty was in the scattered wave, muffin-tin implementation of the time. Major numerical improvements came about through the introduction of Gaussian-type orbitals (GTO) and the use of auxiliary fitting functions in the LCAO-X
method had been tried and abandoned by quantum chemists. Part of the difficulty was in the scattered wave, muffin-tin implementation of the time. Major numerical improvements came about through the introduction of Gaussian-type orbitals (GTO) and the use of auxiliary fitting functions in the LCAO-X![]() program [9,10]. This allowed GTO technology
to be borrowed from existant ab initio codes. Other major advances
were Axel Becke's introduction of efficient, accurate, atom-centered numerical
integration, Vosko, Wilk and Nusair's parameterization of the local density
approximation [11] based upon Ceperly and Alder's accurate quantum Monte Carlo calculations of the correlation energy of the homogeneous electron gas, as well as the emergence of good quality generalized gradient approximations such as those of Becke [12] and Perdew [13].
By the 1980's, it had become clear that it was time to update the old LCAO-X
program [9,10]. This allowed GTO technology
to be borrowed from existant ab initio codes. Other major advances
were Axel Becke's introduction of efficient, accurate, atom-centered numerical
integration, Vosko, Wilk and Nusair's parameterization of the local density
approximation [11] based upon Ceperly and Alder's accurate quantum Monte Carlo calculations of the correlation energy of the homogeneous electron gas, as well as the emergence of good quality generalized gradient approximations such as those of Becke [12] and Perdew [13].
By the 1980's, it had become clear that it was time to update the old LCAO-X![]() strategy and write a modern DFT program with analytic derivatives capable of automatic geometry optimizations. This goal was realized simultaneously in deMon and in another program (namely DGauss developed at Cray for use on their computers).
strategy and write a modern DFT program with analytic derivatives capable of automatic geometry optimizations. This goal was realized simultaneously in deMon and in another program (namely DGauss developed at Cray for use on their computers).
Shortly after its appearance the original deMon code was substantially modified for commercialization by BIOSYM Technologies. The beta-release of this version appeared in 1993. It was the basis of the deMon-KS1 [14] series of programs developed in Montreal until 1997. Meanwhile the original deMon version was further developed in Montpellier and Stockholm. These developments were initially independent from each other. In 1997 they merged to become the deMon-KS3 [15] series of programs.
Over the years, many important method developments in DFT can be attributed to work originating with deMon and related programs. This includes, but is not limited to, NMR chemical shifts [16] and time-dependent density functional theory [17]. Several different versions of deMon developed.
The
The deMon and
|
|
| Last Update: 1st July 2024 | Webmaster |