Next: Keyword ORBITALS Up: SCF Control and Stabilization Previous: SCF Control and Stabilization Contents
| RKS | The restricted Kohn-Sham method will be used. This is the default for closed-shell systems. | ||
| UKS | The unrestricted Kohn-Sham method will be used. This is the default for open-shell systems. | ||
| ROKS | The spin-restricted open-shell Kohn-Sham method will be used. | ||
|
| The ROKS option string defines the ROKS parametrization according to Table 8. By default the Guest & Saunders parametrization [174] is used. | ||
| CUKS | The constrained unrestricted Kohn-Sham method will be used. | ||
|
MAX= | Maximum number of SCF iterations. Default is 100. | ||
|
TOL= | MinMax SCF energy convergence criterion. Default is
| ||
|
CDF= | Auxiliary density convergence criterion. Default
is
| ||
| NOTIGHTEN | The SCF convergence criteria will not be adjusted during an optimization, frequency analysis or property calculation. |
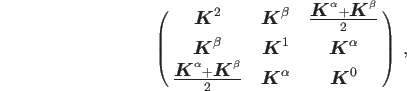 |
(12) |
| (13) | |||
| (14) | |||
| (15) |
Note that the ROKS orbital energies vary with respect to these parametrizations and,
therefore, are not uniquely defined. In particular, the Aufbau principle for the
doubly and singly occupied molecular orbital energy manifolds can be violated
[179,180]. Moreover, ROKS orbital energies include additional constraints
and cannot be compared with RKS or UKS orbital energies. Specifically, the
Slater-Janak theorem [181,182] does not hold for ROKS calculations. For
this reason the block-diagonalized ROKS ![]() and
and ![]() MO energies and
coefficients [183], also named semi-canonical spin orbitals, are printed
after the ROKS energy as shown in the following example output for a
B3LYP/STO-3G/GEN-A2* calculation of the triplet O
MO energies and
coefficients [183], also named semi-canonical spin orbitals, are printed
after the ROKS energy as shown in the following example output for a
B3LYP/STO-3G/GEN-A2* calculation of the triplet O![]() ground state.
ground state.
*** SCF CONVERGED ***
MO COEFFICIENTS OF CYCLE 7
7 8 9 10
-0.3794 -0.0601 -0.0601 0.4267
2.0000 1.0000 1.0000 0.0000
1 1 O 1s 0.0000 0.0000 0.0000 -0.0917
2 1 O 2s 0.0000 0.0000 0.0000 0.5671
3 1 O 2py 0.6576 0.0822 0.7637 0.0000
4 1 O 2pz 0.0000 0.0000 0.0000 0.9484
5 1 O 2px 0.0374 0.7637 -0.0822 0.0000
6 2 O 1s 0.0000 0.0000 0.0000 0.0917
7 2 O 2s 0.0000 0.0000 0.0000 -0.5671
8 2 O 2py 0.6576 -0.0822 -0.7637 0.0000
9 2 O 2pz 0.0000 0.0000 0.0000 0.9484
10 2 O 2px 0.0374 -0.7637 0.0822 0.0000
RANDOMIZED SCF GRID GENERATED IN 3 CYCLES
REFERENCE VALUE OF S**2 FOR PURE SPIN STATE S(S+1): 2.0000
S**2 BEFORE SPIN PROJECTION: 2.0000
S**2 AFTER SPIN PROJECTION: 2.0000
ELECTRONIC CORE ENERGY = -260.328412563
ELECTRONIC COULOMB ENERGY = 101.099784250
ELECTRONIC HARTREE ENERGY = -159.228628313
EXCHANGE ENERGY = -16.350046036
CORRELATION ENERGY = -0.733801619
EXCHANGE-CORRELATION ENERGY = -17.083847655
ELECTRONIC SCF ENERGY = -176.312475968
NUCLEAR-REPULSION ENERGY = 28.059106307
TOTAL ENERGY = -148.253369660
BLOCK DIAGONAL ALPHA MO COEFFICIENTS
7 8 9 10
-0.4028 -0.1618 -0.1618 0.4020
1.0000 1.0000 1.0000 0.0000
1 1 O 1s 0.0729 0.0000 0.0000 -0.0917
2 1 O 2s -0.3516 0.0000 0.0000 0.5671
3 1 O 2py 0.0000 -0.7148 -0.2813 0.0000
4 1 O 2pz 0.6083 0.0000 0.0000 0.9484
5 1 O 2px 0.0000 0.2813 -0.7148 0.0000
6 2 O 1s 0.0729 0.0000 0.0000 0.0917
7 2 O 2s -0.3516 0.0000 0.0000 -0.5671
8 2 O 2py 0.0000 0.7148 0.2813 0.0000
9 2 O 2pz -0.6083 0.0000 0.0000 0.9484
10 2 O 2px 0.0000 -0.2813 0.7148 0.0000
BLOCK DIAGONAL BETA MO COEFFICIENTS
7 8 9 10
-0.3239 0.0415 0.0415 0.4515
1.0000 0.0000 0.0000 0.0000
1 1 O 1s 0.0000 0.0000 0.0000 -0.0917
2 1 O 2s 0.0000 0.0000 0.0000 0.5671
3 1 O 2py -0.3948 -0.7637 -0.0825 0.0000
4 1 O 2pz 0.0000 0.0000 0.0000 0.9484
5 1 O 2px 0.5272 0.0825 -0.7637 0.0000
6 2 O 1s 0.0000 0.0000 0.0000 0.0917
7 2 O 2s 0.0000 0.0000 0.0000 -0.5671
8 2 O 2py -0.3948 0.7637 0.0825 0.0000
9 2 O 2pz 0.0000 0.0000 0.0000 0.9484
10 2 O 2px 0.5272 -0.0825 0.7637 0.0000
The printing of the MO energies and coefficients is activated with
PRINT MOS=7-10 (see Section 4.12.2). The block diagonal ![]() and
and
![]() MO energies and coefficients can be directly compared with their UKS
counterparts. In deMon2k they are also used for ROKS perturbation theory
calculations. As an alternative to ROKS the constrained unrestricted
Kohn-Sham (CUKS) method can be used for spin-projection [184] where the
semi-canonical orbitals are obtained directly. The convergence behavior of
ROKS and CUKS calculations is generally different and it is advisable to
switch among them to test convergence in problematic cases.
MO energies and coefficients can be directly compared with their UKS
counterparts. In deMon2k they are also used for ROKS perturbation theory
calculations. As an alternative to ROKS the constrained unrestricted
Kohn-Sham (CUKS) method can be used for spin-projection [184] where the
semi-canonical orbitals are obtained directly. The convergence behavior of
ROKS and CUKS calculations is generally different and it is advisable to
switch among them to test convergence in problematic cases.
As a consequence of the variational fitting of the density [5,6], deMon2k can exploit a MinMax SCF procedure [185]. In a variational MinMax procedure, there is no strict convergence from above. Therefore, it is possible to obtain energies below the converged energy during the SCF iterations in deMon2k. Note that the printed SCF ERROR for an SCF cycle is the difference between upper and lower MinMax energy bounds and, thus, a direct measurement of how far the current SCF step is away from the convergence point.
The maximum number of SCF iterations is specified with the MAX option. With MAX=0 an "energy only" calculation with the molecular orbital coefficients from the restart file can be performed. No SCF iteration is done! The MAX=0 option automatically triggers GUESS RESTART (see 4.5.5) and, therefore, fails if no adequate restart file deMon.rst exists. The ordering and, thus, the occupation of the molecular orbitals in the restart file can be altered with the MOEXCHANGE keyword (see Section 4.4.3).
The SCF energy convergence criterion can be defined by the user with the TOL
option. Such a user-defined SCF convergence criterion is valid for the first
single-point SCF calculation. During a geometry optimization, however, the
convergence criterion is automatically tightened according to the residual
forces (see Table 9). However, if the user-defined convergence
criterion is smaller than the automatic-tightening value, the user-defined
value is used instead. If self-consistent perturbation calculations are performed,
the automatically requested SCF energy convergence criterion is ![]() Hartree.
It too may be overridden by a smaller value with the TOL option.
Hartree.
It too may be overridden by a smaller value with the TOL option.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The auxiliary density convergence criterion can be defined by the user with the
CDF option. As with the user-defined energy convergence criterion, the user-defined
auxiliary density convergence criterion holds for the first single-point SCF
calculation and overrides the automatically determined CDF values (see Table
9) during the optimization if the user-defined value is smaller.
If self-consistent perturbation calculations are performed, the default auxiliary
density convergence criterion is tightened to
![]() . Again, this value
can be overridden by a smaller user-defined value with the CDF option. At SCF
convergence both energy and auxiliary density convergence criteria are satisfied.
. Again, this value
can be overridden by a smaller user-defined value with the CDF option. At SCF
convergence both energy and auxiliary density convergence criteria are satisfied.
The option NOTIGHTEN disables the automatic tightening of the SCF convergence criteria according to the root mean square (RMS) gradient (as shown in Table 9). Thus, the SCF convergence tolerance is relaxed during geometry optimization and, therefore, SCF convergence failures are less likely. However, accuracy of the gradients is compromised and the structural optimization may not converge. Good practice, therefore, is to use NOTIGHTEN only at the beginning of the optimization or in combination with carefully tested user-defined TOL and CDF values. The NOTIGHTEN option also disables tightening of SCF convergence for perturbation calculations.