Next: Keyword AUGMENT Up: Atom Specific Input Previous: Atom Specific Input Contents
|
| The basis set string |
![]() atomic symbol
atomic symbol![]()
![]() element symbol
element symbol![]()
![]() global basis set
global basis set![]()
Thus any basis set definition for an atom can be overridden by
the explicit assignment of the basis set using the atomic symbol. For example,
for C![]() H
H![]() (see input in 4.1.1), the following basis set definition
(see input in 4.1.1), the following basis set definition
Basis (DZVP) C (TZVP) C1 (STO-3G)
assigns an STO-3G basis to atom C1, a TZVP basis to the other carbon and the DZVP basis to all other atoms. Instead of using basis set strings, the basis set of an atom may also be specified by the Huzinaga notation given in the BASIS file. In that notation, the foregoing basis set definition would read
Basis (DZVP) C (7111/411/1*) C1 (33/3)
The file BASIS contains the basis sets listed in Table 7. Other basis sets can be obtained from the Extensible Computational Chemistry Environment Basis Set Database [131] at www.basissetexchange.org by choosing the deMon2k basis set format.
|
Instead of reading the basis set from the BASIS file, the user can define the basis directly in the input file according to the format:
Basis
SYMBOL Read
N L K
EXPONENT COEFFICIENT
: :
EXPONENT COEFFICIENT
Here SYMBOL can be an element (e.g. OXYGEN) or atomic symbol
(e.g. O), N and L are the principal and angular momentum quantum
numbers of this shell and K
is the degree of contraction. A shell collects all contracted orbitals
of the same angular momentum quantum number, such as ![]() ,
, ![]() ,
, ![]() or
or
![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() and
and ![]() . The contracted
orbitals,
. The contracted
orbitals, ![]() , are linear combinations of (atom-centered) Gaussian
type orbitals (LCGTO), which are called the primitive orbitals,
, are linear combinations of (atom-centered) Gaussian
type orbitals (LCGTO), which are called the primitive orbitals,
![]() :
:
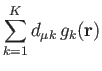 |
(5) | ||
| (6) |
Multiplicity 3 SCFType ROKS VxcType Basis BLYP Mixing 0.4 PRINT GTO CGTO AUXIS Geometry Z-Matrix O O O r # Variables r 1.207 BASIS O Read 1 0 2 49.98097100 0.4301280000 8.896588000 0.6789140000 2 0 2 1.945237000 0.4947200000E-01 0.493363000 0.9637820000 2 1 2 1.945237000 0.5115410000 0.493363000 0.6128200000 AUXIS (GEN-A2)
In this example the basis set was taken from
www.basissetexchange.org
using the deMon2k format (see Figure 6). The first line
in this format (here 3) must be deleted in the user-defined basis set input.
This line defines the number of contractions and is only needed for the basis
set definition in the file BASIS. The basis set in this example contains
a ![]() ,
, ![]() and
and ![]() shell. Each shell has a contraction degree of two. The
shell. Each shell has a contraction degree of two. The
![]() and
and ![]() shells share a common set of exponents. However, in deMon2k both
those shells have to be listed independently, as shown here.
shells share a common set of exponents. However, in deMon2k both
those shells have to be listed independently, as shown here.
The specification of ECP and MCP valence basis sets will also trigger the use of
the corresponding effective or model core potential if the ECP and MCP keywords
are not specified. Thus, their specification usually is not necessary.
An exception is the Hay-Wadt valence basis sets (ECP![]() HW and QECP
HW and QECP![]() HW), which
represent alternative choices to the corresponding LANL (Los Alamos National
Laboratory) double
HW), which
represent alternative choices to the corresponding LANL (Los Alamos National
Laboratory) double ![]() valence basis sets. If the Hay-Wadt basis sets are
used, the ECP must be specified explicitly with the ECPS keyword. This is a
special case of the more general capability of defining basis sets and ECPs
or MCPs independently by the BASIS and ECPS or MCPS keywords. Be aware that
you can mix any ECPs/MCPs with any basis set, including all-electron ones! Thus,
care must be taken if the keywords BASIS and ECPS/MCPS are used together in the
deMon2k input. For less experienced users, we recommend using the PRINT keyword
with the BASIS options ECPS and MCPS (see example
valence basis sets. If the Hay-Wadt basis sets are
used, the ECP must be specified explicitly with the ECPS keyword. This is a
special case of the more general capability of defining basis sets and ECPs
or MCPs independently by the BASIS and ECPS or MCPS keywords. Be aware that
you can mix any ECPs/MCPs with any basis set, including all-electron ones! Thus,
care must be taken if the keywords BASIS and ECPS/MCPS are used together in the
deMon2k input. For less experienced users, we recommend using the PRINT keyword
with the BASIS options ECPS and MCPS (see example ![[*]](crossref.png) on page
of the tutorial) in order to obtain full information about the basis sets and
ECPs/MCPs actually utilized. If the ECP or MCP valence basis sets are obtained
from external resources like www.basissetexchange.org, the principal
quantum number indexing may be wrong. As a result the tight-binding start density
cannot be generated correctly. As a quick fix, we suggest switching to a CORE start
density by GUESS CORE (see 4.5.5).
on page
of the tutorial) in order to obtain full information about the basis sets and
ECPs/MCPs actually utilized. If the ECP or MCP valence basis sets are obtained
from external resources like www.basissetexchange.org, the principal
quantum number indexing may be wrong. As a result the tight-binding start density
cannot be generated correctly. As a quick fix, we suggest switching to a CORE start
density by GUESS CORE (see 4.5.5).
![\includegraphics[width=13.0cm]{/home/gerald/guide.5.0/Figures.5.0/Basis_Set_STO-2G.eps}](ug-img189.png)